Updated! 1969 Big Bertha Paint Scheme on a Super Big Bertha.
By Scott Johnson
2021-07-31
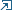
I redid the 1969 Big Bertha paint scheme with the following updates.
Changed stripes to 2cm wide instead of 2.3cm.
Shortened barber pole to 9cm long.
Lowered the bottom most black stripe by 2cm thus providing more center white space.
I think these changes make the model look closer to the Ninfinger Productions: 1969 Estes Catalog picture.
Image to the left is my first try and image to the right is the updated paint scheme.
* Note how the barber pole stripes land vertically over each other on the first try and on the second they don't.
** in the actual 1969 Big Bertha paint scheme the stripes may be closer to 1.75cm for the Super Big Bertha - compared to the catalog image, but I'm done with this bird.


Still pending the logo... Will get that done soon enough.
This one minus the writing and arrow around the center black and white logo - see Nine Fingers catalog.

Here is an image of the 9 hour clock (described below) to create the barber pole.

Original build:
-----------------------------------------------------------------------------------------------------------------------------------
1969 Big Bertha Paint Scheme on a Super Big Bertha.
Ninfinger Productions: 1969 Estes Catalog
I did this by eye balling images of this paint scheme and I do not have the official method.
I would be every interested if an official method to paint this scheme exists.

The trick for the 1969 Big Bertha is that the width of the stripes on the rocket are 1/9th the circumference of the body tube. That’s it!
For example:
The Super Big Bertha is a 2.6” diameter rocket (painted it’s 2.61”).
Circumference = 8.16814” (add a little for paint 8.2”)
8.2/9=.911” stripes
*I actually did mine in centimeters and had 2.32cm stripes.
For the barber pole effect you have three red stripes and each stripe is 1/9 of the circumference.
So it's one stripe and two blanks, one stripe and two blanks, one stripe and two blanks.
The length of the barber pole, on my rocket, is 5", but I bet it's actually something like 4" or 4 stripe widths long...?
I marked every 2.32cm around the body tube where the black bands (top and bottom) are that make up the barber pole, like a nine hour clock top and bottom.
The stripes wrap from a top mark and count around the base four marks. It may actually be count five marks around to find where the bottom stripe should land, but I was happy with four.
I laid my stripes down with electrical tape because I’m a cheater. 😉
I've found most people never look close enough to tell the difference (but this is my biggest cheat to date) and no one ever really sees your shortcuts (or mistakes) unless you point them out.
Electrical or pin-striping tape does fine on LPR rockets, but that's as far as I would take it.
I may swing back and paint this rocket for real as I now have all the outlines for my masking lines already laid out. It would definitely hold up better to close up inspection if it were painted.
Before that I'll implement the fixes with tape and post them here - reduce barber pole to 4 stripe widths long and wrap red barber pole stripes 4 marks around on the 9 hour clock - to see if I can get it a bit closer to the catalog image.
I just need to lift and adjust the tape, but that's for another time (actually I have marks under tape so I would probably repaint the white top before any changes).
For the red electrical tape, I painted the electrical tape with the same red paint used on the body of the rocket. Be sure to lightly scratch up the electrical tape with 400 grit sandpaper before painting.
You need a deeper red for this rocket, and I went with Rust-Oleum Gloss Protective Enamel Regal Red

Pictures are in direct sun so it looks a lighter red than it really is, I'll try and post a image that is more true to it's color.
Still need to order or make the Estes logo

 |
 |